
Cystic Fibrosis Patients-Patient Registry Report
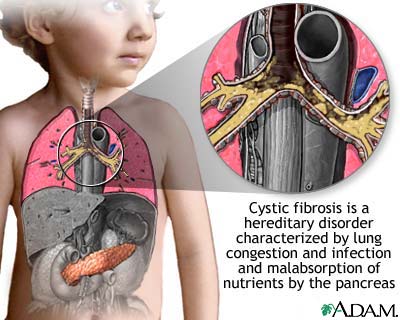
Cystic fibrosis (also known as CF or mucoviscidosis) is an autosomal recessive ... Ciliated epithelial cells in the patient have a mutated protein

More than 40 years ago, the CF Foundation created the Cystic Fibrosis Patient Registry to track the health of people with CF in the United States.
The information in this registry allows health care professionals and researchers to identify new health trends, recognize the most effective treatments and design clinical trials for potential therapies.
The registry anonymously reports patient data from more than 27,000 CF patients who receive care at CF Foundation-accredited centers and agree to participate in the registry.
This data also can be used to find areas where more work can be done to improve the health of those with the disease.
It isn’t easy to cure a genetic disease. For the luckier patients, robust therapies help them manage the symptoms and avoid complications like infections. But in the end, the only way to fix an inherited disorder like cystic fibrosis, which affects some 70,000 children and adults worldwide with its life-threatening buildup of thickened mucus, is to repair the fundamental building blocks of DNA that went astray.
 In the 1950s, patients who battled the recurrent infections of cystic fibrosis often suffered from malnutrition and rarely lived past kindergarten. By the 1980s, despite newly available medications, most still only made it to their high school graduation.
In the 1950s, patients who battled the recurrent infections of cystic fibrosis often suffered from malnutrition and rarely lived past kindergarten. By the 1980s, despite newly available medications, most still only made it to their high school graduation.
But today, thanks to novel therapies, specialized care centers and newborn screening programs, many patients are living well into middle age. The median age of survival now exceeds 37, according to the Cystic Fibrosis Foundation. Experts say that figure will likely continue to increase with the development of new drugs that could make cystic fibrosis a controlled disease like asthma — or even cure it.
“It’s not the same disease we saw 15 years ago,” said Dr. Carlos Milla, an associate professor at Lucile Packard Children’s Hospital at Stanford University. He has noticed a shift from seeing very sick children to very sick adults. “The outlook for patients with cystic fibrosis has changed dramatically,” he added. “There are a number of therapies on the horizon that will either lead to a cure or a very definitive control of the disease.”
That shift, physicians say, stems largely from more effective medications, introduced over the last 15 years, that combat the thick mucus that builds up inside the body and impairs vital organs like the lungs and pancreas.
“It is like oatmeal that has been left on the stove too long,” said Dr. Preston Campbell III, a pediatric pulmonologist at Johns Hopkins Hospital in Baltimore. “Their mucus becomes difficult to clear and, as a result, the lungs get obstructed, infected and inflamed.”
A new string of inhaled medications makes it easier for patients to clear out the airways and, ultimately, avoid infection. Pulmozyme, which came out in 1994, makes the mucus thinner and easier to expel. A few years later, the Food and Drug Administration approved inhaled tobramycin, an antibiotic that delivers medication straight into the lungs. The drug provided a better way to fight off infections, including those caused by Pseudomonas bacteria, the most common source of chronic lung infections.
And in 2004 came inhaled hypertonic saline, which helps clear mucus by drawing salt and water back into dehydrated airways. The drug cuts pulmonary flare-ups in half, according to a study in The New England Journal of Medicine, and helped Emily Schaller, a 27-year-old patient from Detroit, recently run a half marathon.
“I started out three years ago not being able to run a block,” said Ms. Schaller, who works in retail. “Crossing that finish line made me realize that I can do whatever I put my mind to. Next up is a full marathon in October.”
“These drugs have represented a quantum leap in care for individuals with cystic fibrosis,” said Dr. James Acton, director of the Cystic Fibrosis Center at Cincinnati Children’s Hospital.
But medications are only one piece of the puzzle. Patients continue to use various airway clearance techniques, which act like chest physical therapy to break up the sticky mucus in the lungs so they can then cough it up.
In addition, over the last two decades the quality of care at centers around the country has continued to improve.
And nearly all states have adopted newborn screening programs to test for the cystic fibrosis gene. Research has shown that early detection, particularly in infancy, can help prevent both pulmonary and gastrointestinal complications later in life.
Bill Elder, 21, who has to spend nearly two hours a day clearing his lungs, is certain that early testing would have helped him. He went eight years without a diagnosis and, more important, without treatment, because he was born in Nebraska, which started testing infants in 2006. “It should be nationwide,” said Mr. Elder, a Stanford University undergraduate.
Researchers agree there is still a lot of work to do. “We need to focus not only on developing therapies that treat the downstream effects of disease but also on the therapies that are further upstream — those that can intervene early and either improve the complications that arise or prevent them entirely,” Dr. Acton said.
Twenty years ago, researchers thought they were on the cusp of such a discovery after scientists located the faulty gene that causes the disease. It was a huge breakthrough, raising hopes that investigators might be able to use gene therapy — a method that delivers healthy genes back into the body’s cells and tissues — to find a cure.
But that line of research simply didn’t pay off.
Today, physicians say the most exciting avenue of research involve new drugs that could address the root of the problem.
Over the last decade, for example, scientists have been testing an experimental compound called VX-770, which may be able to fix the defective protein that causes the surface of the lungs to become so dehydrated in cystic fibrosis. It works by essentially tricking the cells that line the airway into secreting salt, which causes water to flow back into the airway. By restoring the proper balance of salt and water, the lungs become hydrated, the mucus thins out and harmful bacteria are more easily washed away.
“I do think we will see a cure,” said Dr. Richard Boucher, director of the Cystic Fibrosis/Pulmonary Research and Treatment Center at the University of North Carolina at Chapel Hill. “These therapies that hydrate the cystic fibrosis airway surface may be able to stop the progression of the disease in adults. And, very excitingly, in babies you may even be able to prevent it.”
Dr. Boucher says the likelihood that this will happen in the next 5 to 10 years is “extremely high.” But many scientists also warn that even the most promising of drugs can fail in late-stage testing. There are now 30 or so drugs in the pipeline, including VX-809, another compound that helps correct the defective cystic fibrosis gene. If one fails, another might make it through eventually.
Cystic fibrosis (also known as CF or mucoviscidosis) is an autosomal recessive ... Ciliated epithelial cells in the patient have a mutated protein

More than 40 years ago, the CF Foundation created the Cystic Fibrosis Patient Registry to track the health of people with CF in the United States.
The information in this registry allows health care professionals and researchers to identify new health trends, recognize the most effective treatments and design clinical trials for potential therapies.
The registry anonymously reports patient data from more than 27,000 CF patients who receive care at CF Foundation-accredited centers and agree to participate in the registry.
This data also can be used to find areas where more work can be done to improve the health of those with the disease.
It isn’t easy to cure a genetic disease. For the luckier patients, robust therapies help them manage the symptoms and avoid complications like infections. But in the end, the only way to fix an inherited disorder like cystic fibrosis, which affects some 70,000 children and adults worldwide with its life-threatening buildup of thickened mucus, is to repair the fundamental building blocks of DNA that went astray.
 In the 1950s, patients who battled the recurrent infections of cystic fibrosis often suffered from malnutrition and rarely lived past kindergarten. By the 1980s, despite newly available medications, most still only made it to their high school graduation.
In the 1950s, patients who battled the recurrent infections of cystic fibrosis often suffered from malnutrition and rarely lived past kindergarten. By the 1980s, despite newly available medications, most still only made it to their high school graduation.But today, thanks to novel therapies, specialized care centers and newborn screening programs, many patients are living well into middle age. The median age of survival now exceeds 37, according to the Cystic Fibrosis Foundation. Experts say that figure will likely continue to increase with the development of new drugs that could make cystic fibrosis a controlled disease like asthma — or even cure it.
“It’s not the same disease we saw 15 years ago,” said Dr. Carlos Milla, an associate professor at Lucile Packard Children’s Hospital at Stanford University. He has noticed a shift from seeing very sick children to very sick adults. “The outlook for patients with cystic fibrosis has changed dramatically,” he added. “There are a number of therapies on the horizon that will either lead to a cure or a very definitive control of the disease.”
That shift, physicians say, stems largely from more effective medications, introduced over the last 15 years, that combat the thick mucus that builds up inside the body and impairs vital organs like the lungs and pancreas.
“It is like oatmeal that has been left on the stove too long,” said Dr. Preston Campbell III, a pediatric pulmonologist at Johns Hopkins Hospital in Baltimore. “Their mucus becomes difficult to clear and, as a result, the lungs get obstructed, infected and inflamed.”
A new string of inhaled medications makes it easier for patients to clear out the airways and, ultimately, avoid infection. Pulmozyme, which came out in 1994, makes the mucus thinner and easier to expel. A few years later, the Food and Drug Administration approved inhaled tobramycin, an antibiotic that delivers medication straight into the lungs. The drug provided a better way to fight off infections, including those caused by Pseudomonas bacteria, the most common source of chronic lung infections.
And in 2004 came inhaled hypertonic saline, which helps clear mucus by drawing salt and water back into dehydrated airways. The drug cuts pulmonary flare-ups in half, according to a study in The New England Journal of Medicine, and helped Emily Schaller, a 27-year-old patient from Detroit, recently run a half marathon.
“I started out three years ago not being able to run a block,” said Ms. Schaller, who works in retail. “Crossing that finish line made me realize that I can do whatever I put my mind to. Next up is a full marathon in October.”
“These drugs have represented a quantum leap in care for individuals with cystic fibrosis,” said Dr. James Acton, director of the Cystic Fibrosis Center at Cincinnati Children’s Hospital.
But medications are only one piece of the puzzle. Patients continue to use various airway clearance techniques, which act like chest physical therapy to break up the sticky mucus in the lungs so they can then cough it up.
In addition, over the last two decades the quality of care at centers around the country has continued to improve.
And nearly all states have adopted newborn screening programs to test for the cystic fibrosis gene. Research has shown that early detection, particularly in infancy, can help prevent both pulmonary and gastrointestinal complications later in life.
Bill Elder, 21, who has to spend nearly two hours a day clearing his lungs, is certain that early testing would have helped him. He went eight years without a diagnosis and, more important, without treatment, because he was born in Nebraska, which started testing infants in 2006. “It should be nationwide,” said Mr. Elder, a Stanford University undergraduate.
Researchers agree there is still a lot of work to do. “We need to focus not only on developing therapies that treat the downstream effects of disease but also on the therapies that are further upstream — those that can intervene early and either improve the complications that arise or prevent them entirely,” Dr. Acton said.
Twenty years ago, researchers thought they were on the cusp of such a discovery after scientists located the faulty gene that causes the disease. It was a huge breakthrough, raising hopes that investigators might be able to use gene therapy — a method that delivers healthy genes back into the body’s cells and tissues — to find a cure.
But that line of research simply didn’t pay off.
Today, physicians say the most exciting avenue of research involve new drugs that could address the root of the problem.
Over the last decade, for example, scientists have been testing an experimental compound called VX-770, which may be able to fix the defective protein that causes the surface of the lungs to become so dehydrated in cystic fibrosis. It works by essentially tricking the cells that line the airway into secreting salt, which causes water to flow back into the airway. By restoring the proper balance of salt and water, the lungs become hydrated, the mucus thins out and harmful bacteria are more easily washed away.
“I do think we will see a cure,” said Dr. Richard Boucher, director of the Cystic Fibrosis/Pulmonary Research and Treatment Center at the University of North Carolina at Chapel Hill. “These therapies that hydrate the cystic fibrosis airway surface may be able to stop the progression of the disease in adults. And, very excitingly, in babies you may even be able to prevent it.”
Dr. Boucher says the likelihood that this will happen in the next 5 to 10 years is “extremely high.” But many scientists also warn that even the most promising of drugs can fail in late-stage testing. There are now 30 or so drugs in the pipeline, including VX-809, another compound that helps correct the defective cystic fibrosis gene. If one fails, another might make it through eventually.
No comments:
Post a Comment